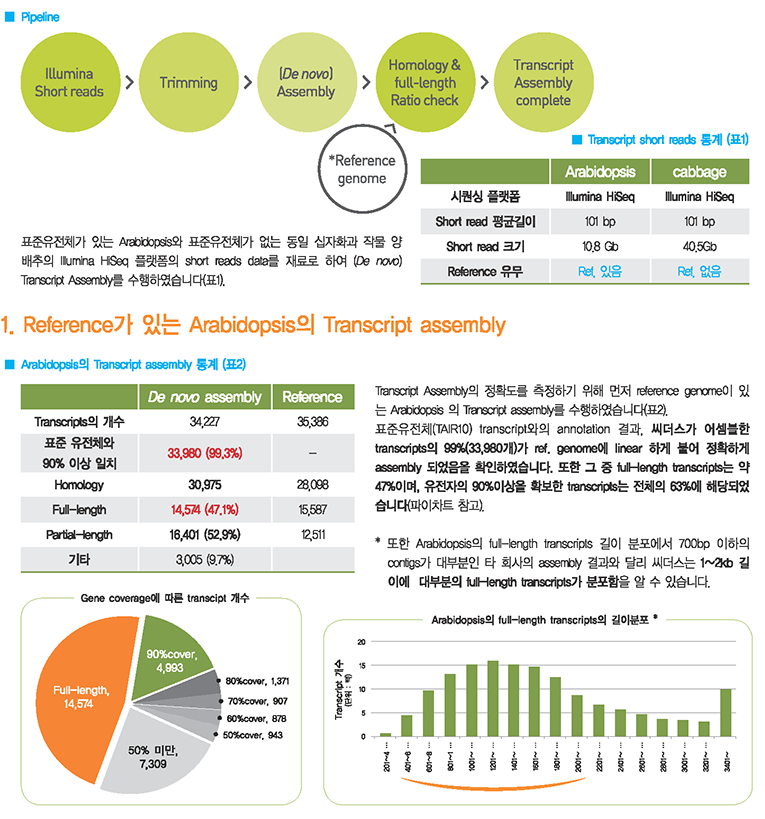
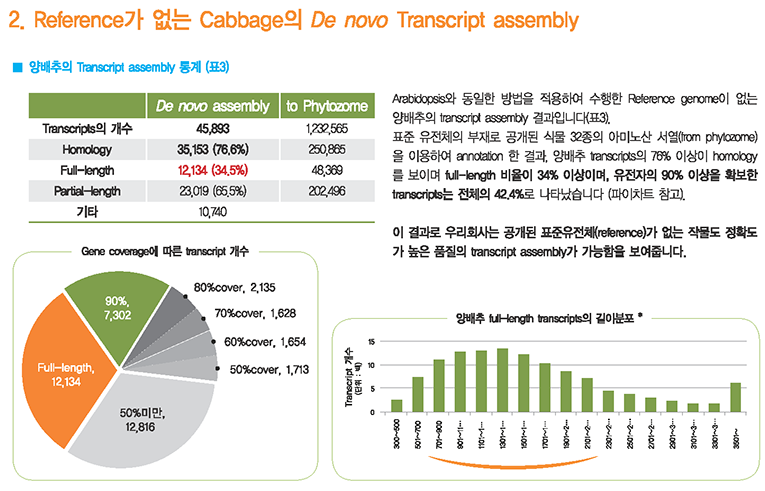
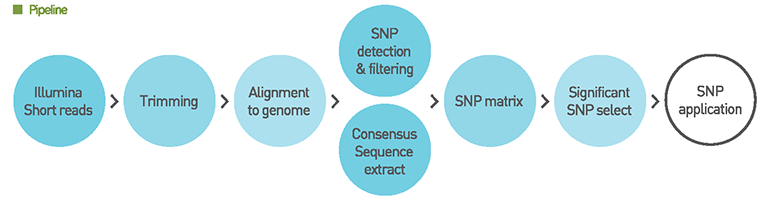
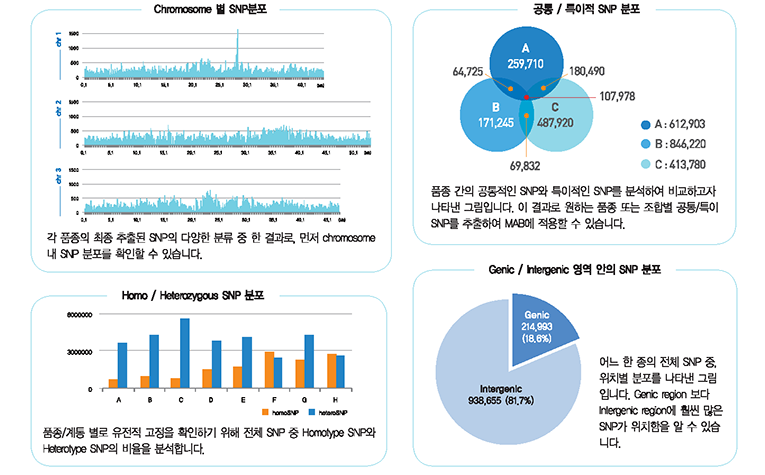
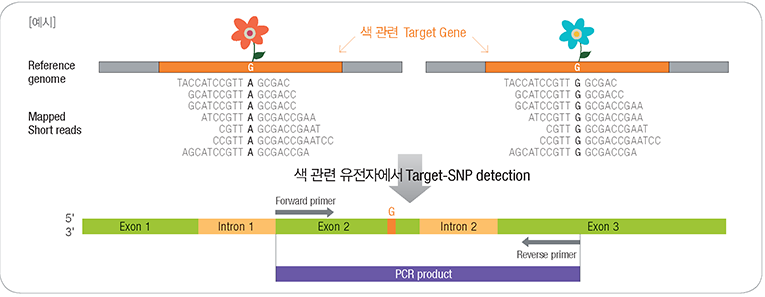
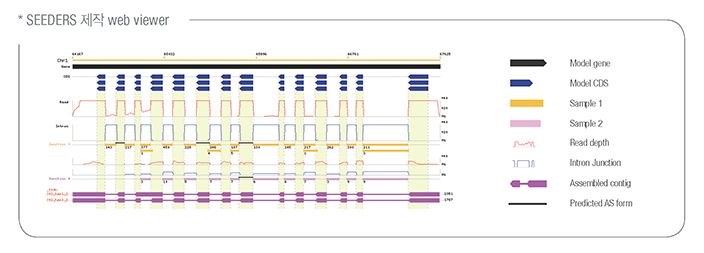
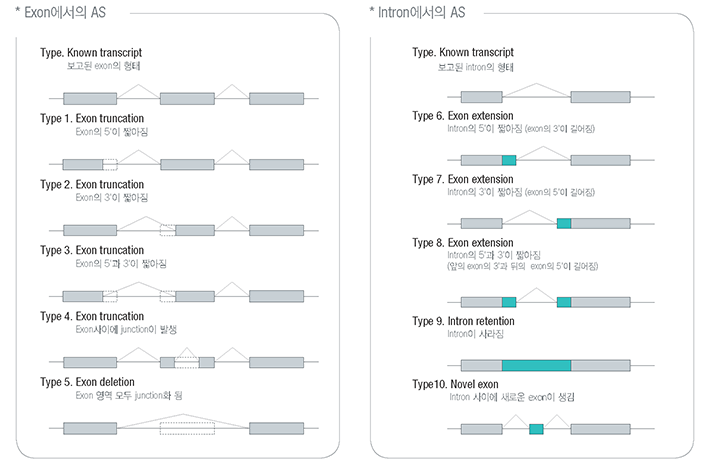
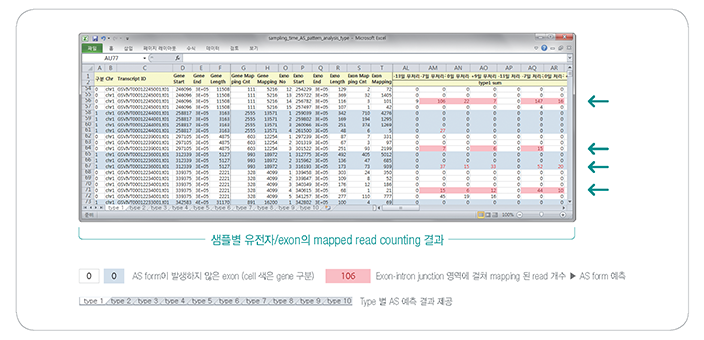
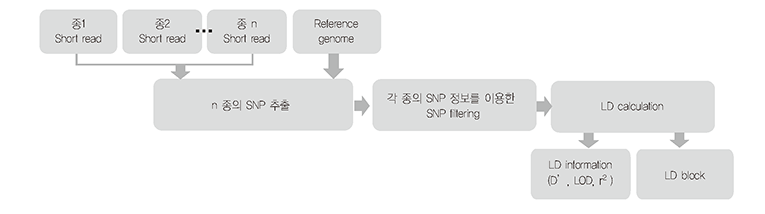
차세대 시퀸싱(NGS)의 비용이 저렴해지면서 교배집단 전체를 시퀀싱하고, 이를 기반으로 한 시퀀스 수준에서의 genotyping이 활발히
이루어지고 있습니다. 이는 기존의 genetic map과는 다르게 수십만개의 마커를 포함하는 초고밀도 genetice map 작성을 목표로 합니다.
이를 통하여 recombination break point등의 유전 양상을 이해할 수 있어 여러 연구에 응용할 수 있습니다.
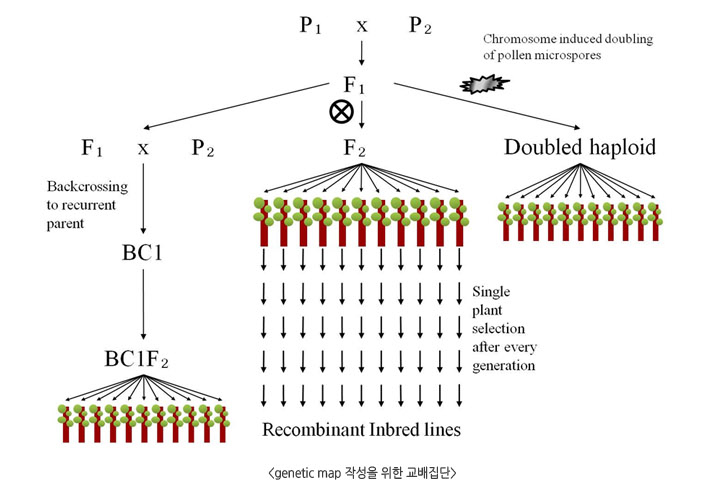
Genetic map을 만들기 위해서는 F2, BC, RIL, DH등 목적에 따라 다양한 집단이 이용되고 있습니다. F2, BC집단은 작성이 단순하고
소요시간이 짧지만 heterozygous한 상태로 임시적인 집단입니다. RIL, DH, NIL은 영속적 집단으로 취급하는데 이는 homozygous하기
때문에 유전적으로 고정된 종자로 증식, 보관이 가능합니다.
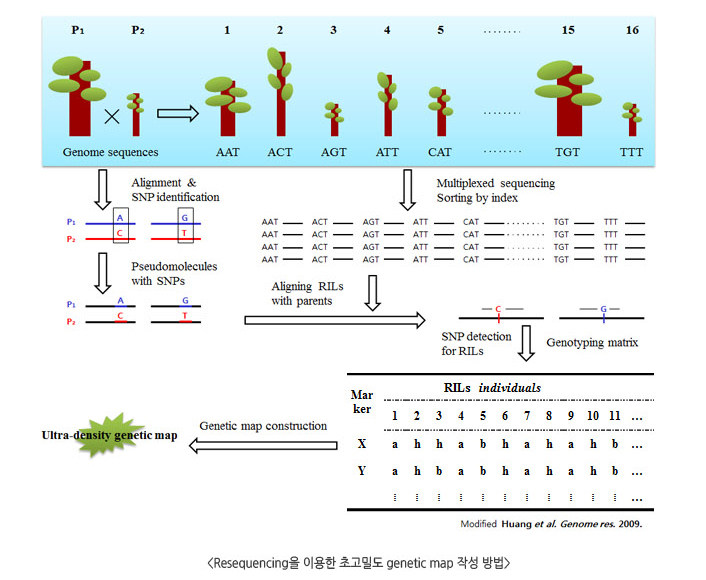
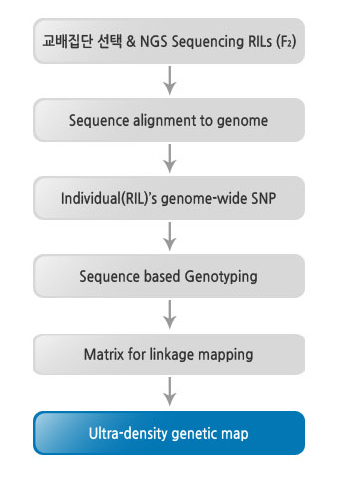
준비된 교배집단을 Multiplexed sequencing 방식을 이용하여 집단전체의 시퀀스를 확보합니다. 시퀸스 기반의 genotyping을 통해 matrix를 작성하고 Ultra-density genetic map을 작성합니다.
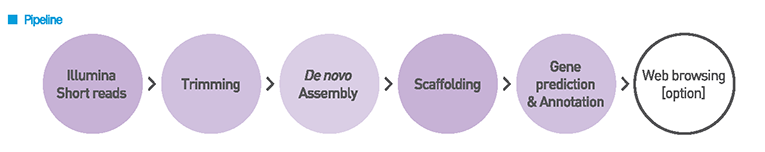
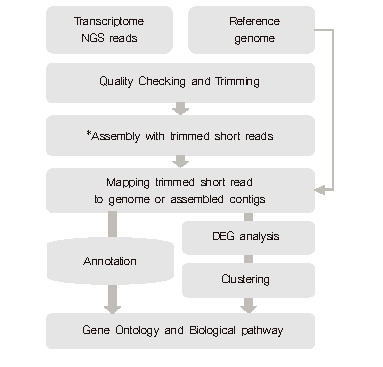
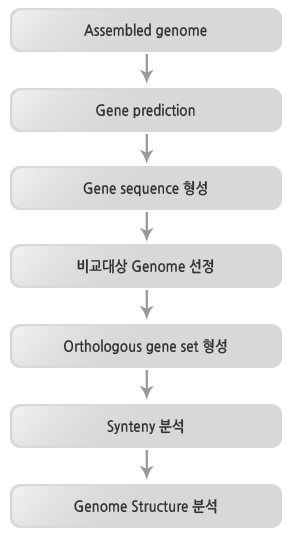
Work Flow
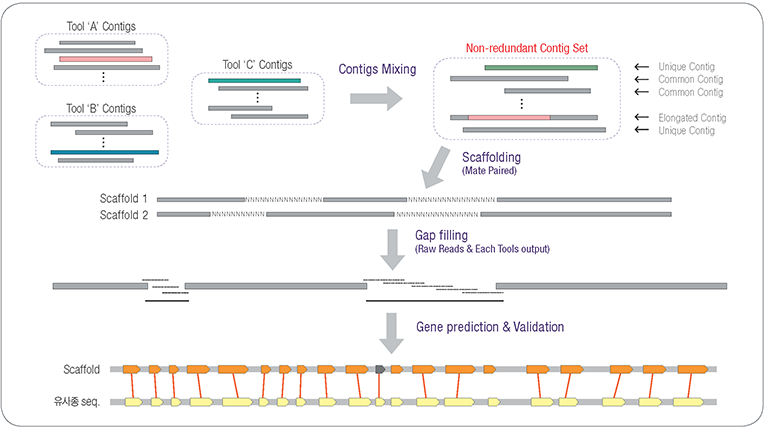
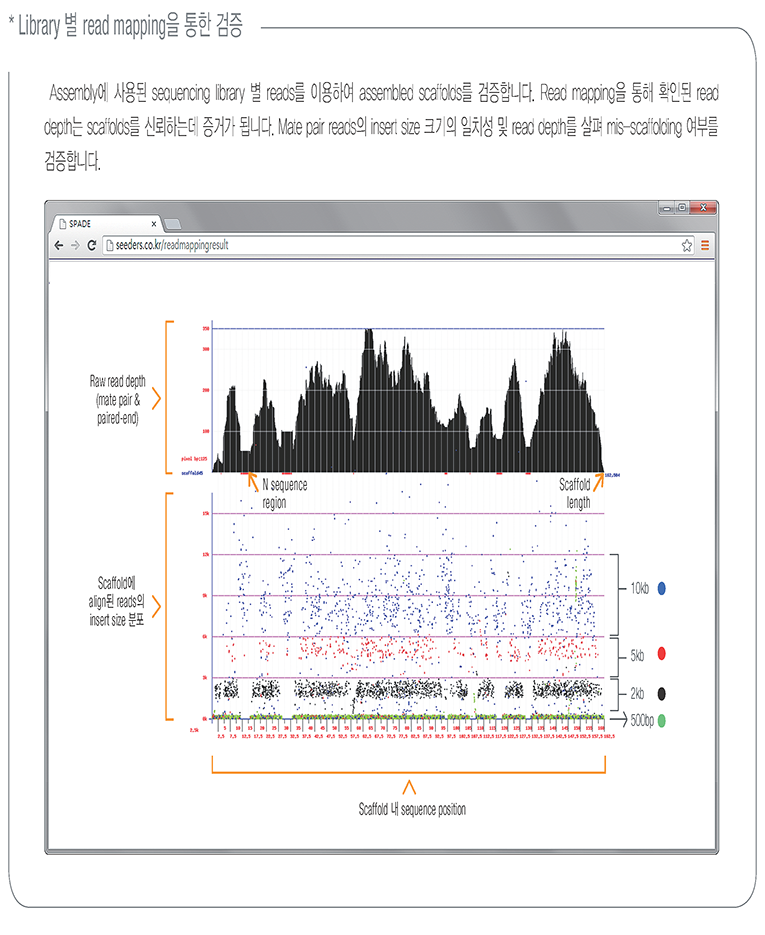
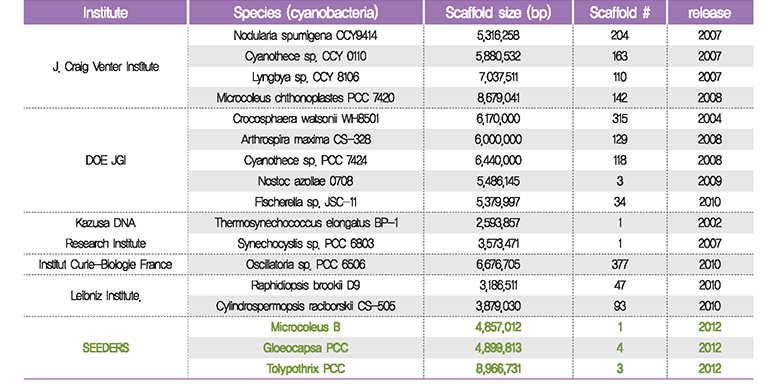
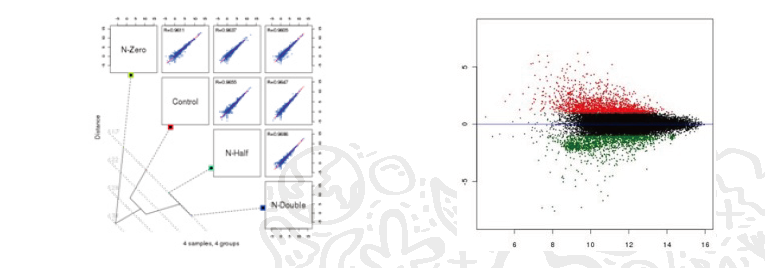
분석과정 및 결과
Low depth로 시퀀싱이 된 경우, 각 개체별 결손되는 SNP가 많이 발생됩니다. 이러한 문제점을 해결하기 위해
알고리즘 개선 및
새로운 프로그램의 개발을 통해 linkage map용 matrix를 획기적으로 개선시킴으로써 high-quality genetic map을 작성
할 수 있습니다.
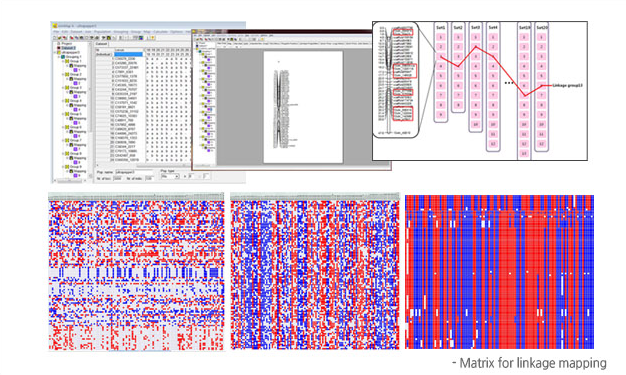
- 100,000 ~ 1,000,000 SNPs 확보가능
- Genomic contig 혹은 scaffold의 순서를 정할 수 있음
- 농업적으로 중요한 유전자 fine mapping & cloning
- 정밀한 지도로 정확한 QTL mapping 가능
- Recombination Break Point를 확인
- Bin map 확보